Artificial intelligence (AI) and machine learning are making a large impact on drug discovery and development. It’s hard to open a copy of C&E News without reading about the latest AI-guided synthetic routes or “smart” models of drug metabolism and toxicology. So what about applying AI to peptides and protein-protein interactions, areas which our lab is heavily invested in? Turns out that there are plenty of opportunities here as well, this is why we recently read about a new machine learning tool dubbed POOL (or Peptide Optimization with Optimal Learning), which is being used to computationally evolve peptides with orthogonal bioactivities. This study, published in Nature Communications, came out of a highly interdisciplinary collaboration between a nanotechnologist, Nathan Gianneschi at Northwestern, a computational chemist, Peter Frazier at Cornell, and chemical biologist, Michael Burkart at UCSD, and resulted in some striking results for their designer peptides.
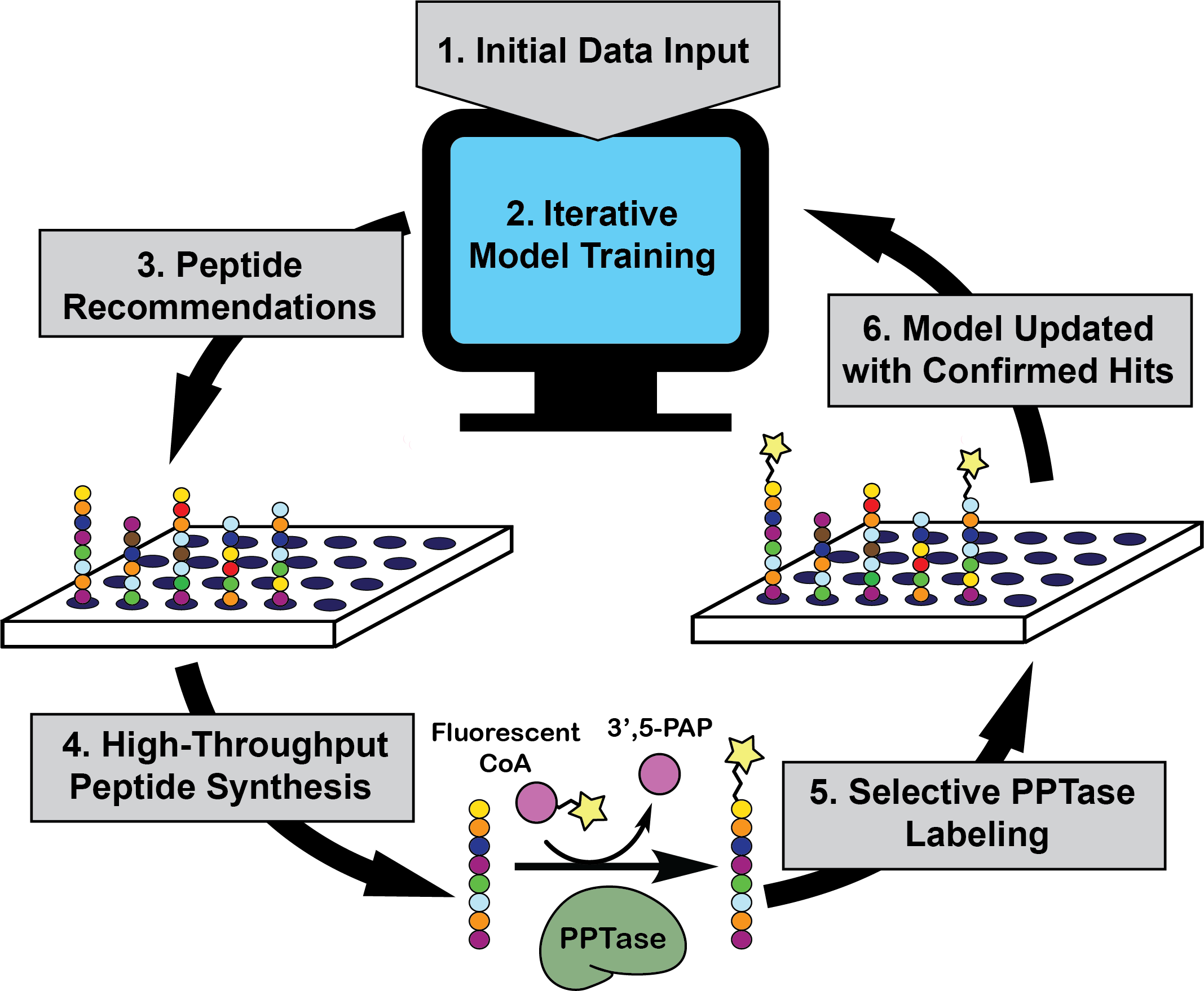
Finding selective, high affinity peptides can be time consuming. POOL uses an initial data set for a given peptide-protein or protein-protein interaction to train its algorithm in predicting new sequences with improved activity or selectivity (Figure 1.1). A quick look “under the hood” finds that POOL’s algorithm exploits a Naïve Bayesian approach to first predict activity for new peptide sequences based on the training set and then select subsets with a large probability of improvement (Figure 1.2). In rounds of the latter step selection, top hits are discarded from the model in order to expand the diversity of sequences being tested. Furthermore, to reduce statistical overfitting, the authors binned the 20 canonical amino acids into eight categories based on their chemical properties. After the algorithm generates an adequate model, the computationally predicted “top-hits” are subjected to experimental validation (Figure 1.3-1.5) and the biochemical results are then fed back into the algorithm to improve future selections (Figure 1.6). By incorporating experimental data and completing multiple iterations, one can improve the model’s ability to reveal peptide sequences that selectively bind to structurally similar enzymes.
In the current study, the authors sought to validate this approach by finding substrates selective for Sfp or Acps, two structurally similar 4’-phosphopantetheinyl transferases (PPTases). PPTases are a superfamily of enzymes that transfer fragments of CoA to the many modular and iterative synthetases used in primary metabolism and biosynthesis; PPTases are used in all forms of life, so selective binders of bacterial or fungal PPTases could be useful antiinfectives and/or chemical probes. The initial training set for POOL was created from three different sources: (1) peptides collected from a phage display selection, (2) known PPTase substrates, and (3) bioinformatically predicted substrates of Sfp and AcpS. Importantly, POOL requires only hundreds of validated peptides to begin predicting new sequences with desired biological activities, rather than the millions employed in similar techniques. SPOT synthesis was used to rapidly prepare the predicted peptide sequences and a fluorescent-CoA assay was used to confirm peptide hits (Figure 1.4-1.5). After just three rounds of predict, validate, and repeat, the algorithm had identified several sequences that proved selective for one of the two PPTase isoforms. Notably, the Sfp-specific sequences all contained hydrophobic residues (Ile/Leu) at positions 2 and 5, consistent with the presence of a structurally defined, non-polar patch on Sfp’s catalytic domain; mutations to these polar residues disrupted contact with the catalytic domain. In contrast, AcpS-selective peptides incorporated polar residues (e.g. Ser, Asp) at these positions. Thus, the sequence diversity imposed by POOL was key in accessing selective binders and helped to probe the structure activity relationships of these interactions.
Overall, we were impressed by the ability of POOL to find de novo peptide substrates for these PPTase enzymes. Several benefits really seemed to stand out. For one, the ability of POOL to simultaneously accommodate design criteria from more than one target stands out over other selection techniques. Whereas in vitro techniques, such as mRNA and phage display, would be technically challenged to simultaneously sample multiple targets in parallel, we could envision POOL quickly multiplexes tens or even hundreds of selection criteria, with the major limiting factor being computing power. Furthermore, the ability to pull training data from genomic sources has profound implications: could this tool be extended to understanding and manipulating the complex interactions that govern NRPS biosynthesis or other protein-protein networks? In addition to probing protein-protein interactions, we wondered whether POOL might be adapted to help overcome some of the limitations associated with peptide drugs, for instance, could the model incorporate solubility or cell permeability along with other selection criteria? Of course, certain aspects of the POOL model could still be improved, such as eliminating the binning of amino acids and increasing complexity of the model. Improving on the categorization of amino acids would enable incorporation of non-canonical amino acids and expand the chemical space applied to some of these questions. POOL is yet another example of how AI could make drug discovery quicker, cheaper, and more effective. We look forward to seeing how the model advances to incorporate some of these additional questions that may exist beyond the capabilities of in vitro screening methods.
One Response to “A Deeper Dive into a Peptide POOL”
Samara
Thanks for sharing this very informative article.