We’ve previously touched on DELs and some of what they’re doing to revolutionize drug discovery. mRNA display is a somewhat similar technique that is increasingly being used to discover new high affinity peptide-based ligands for challenging therapeutic targets. mRNA display takes advantage of synthetic oligonucleotides and cell-free transcription/translation to generate large, naïve libraries of mRNA-barcoded peptides, which can then be used in rounds of selection to search for high affinity binders to a protein target of interest. While the technique was invented in Nobel laureate Jack Szostak’s lab at Harvard Medical School and Mass General Hospital, many major innovations have been made by one of our collaborators, Hiroaki Suga at the University of Tokyo. In particular, the RaPID (or Random nonstandard Peptide Integrated Discovery) platform, developed by Suga’s lab, provides key advances, including: (1) procedural improvements that save time, allowing whole selections inside of a week and (2) incorporation of unnatural amino acids (UAAs) by robust RNA aptamers known as flexizymes. Overall, RaPID has turned mRNA display into an exceptionally powerful tool that is being used to identify potent new peptide inhibitors and has spawned at least one highly successful off-shoot company, PeptiDream.
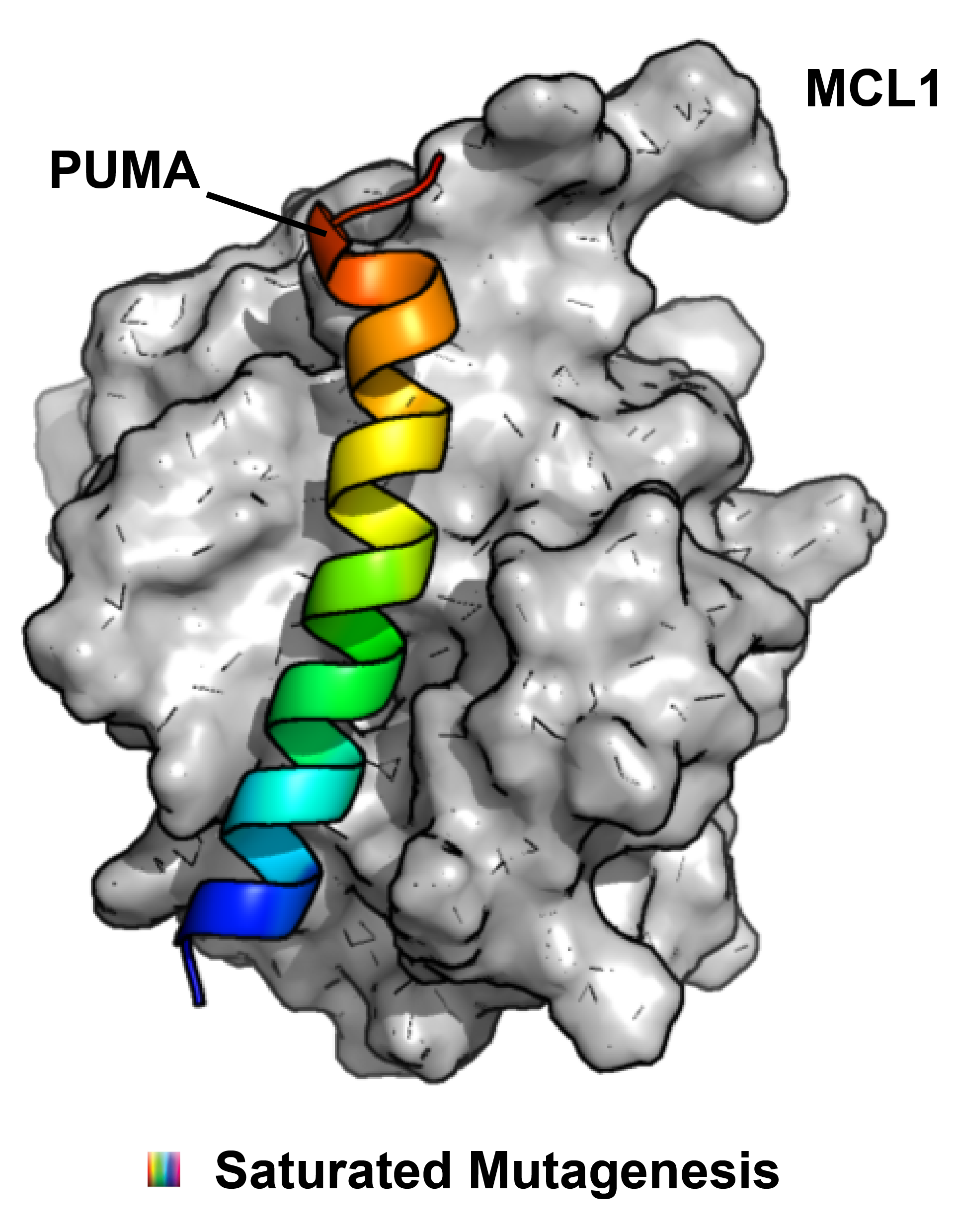
But is mRNA display only able to discover new protein binders, or can it also be used as a hyperfine tool to study existing protein-protein interactions (PPI)? Our lab has been very interested and active in using mRNA display in novel ways and this week we were taking note of a new paper in PNAS from the Suga lab that uses RaPID to study the interaction between the BH3 domain protein PUMA and MCL1. This interaction is a nice place to start because it is already well-studied and the effects of multiple point mutations on ΔΔG of binding are published. mRNA display allowed the authors to scan every position in PUMA with a comprehensive set of single point mutations with 41 different amino acids (20 canonical and 21 unnatural). One key to the experimental design was the validation that ΔΔG correlates with enrichment in single rounds of selection. After some optimization, the calculated and normalized ΔΔGs for known mutants displayed on mRNA could be made to agree linearly with published values. Attendantly, a large PUMA peptide library of single point mutations was prepared using RaPID and the more extensive data set obtained; in this single experiment they were able to gather highly accurate binding data for a large number of mutants, which would have taken years by more traditional methods. In the end, the conclusions were enlightening: importantly, in nature, high affinity does not always lead to optimum fitness. Thus, while many mutations were bad for binding, still many others further stabilized the interaction, especially many of the UAA mutations. For those studying less characterized PPIs or trying to develop PPI inhibitors through rational design, this approach could provide a lot of useful information quickly.
Separately, the authors also analyzed a cyclic peptide, CP-2, which had been identified in a previous mRNA display selection as a high affinity inhibitor of KDM4. Interestingly, very few mutations were stabilizing, indicating that the previous selections had already optimized the affinity of the peptide. Still, although it was hard to find mutations that enhanced the already optimized affinity, several mutations, especially new UAAs, that enhanced overall “drug-like” properties of CP-2 proved neutral, providing a roadmap to potential drug development for this novel inhibitor. Cumulatively, the two sets of experiments, PUMA and CP-2, really elucidate the differences between naturally evolved and artificially selected protein-peptide interactions and what RaPID can teach about the two. On the one hand, this approach has the capacity to quickly unveil the strengths and weakness of a natural interaction and on the other, it also has the potential to rationally build on a new synthetic interaction. Both kinds of insight will have major applications for drug discovery and biophysics.
While this is exciting new research, it is still quite technically challenging and requires a substantial amount of specialized knowledge. For instance, many elements need to be finely tuned before beginning selection, such as: (1) library concentration, (2) immobilized protein concentration, (3) time of library incubation, and (4) sequencing coverage relative to library diversity (for high quality data, every sequence must be counted hundreds to thousands of times). If any of these parameters are not optimized, the data quality and analysis can suffer. Additionally, to incorporate the 21 UAAs, 21 different displayed libraries were prepared and a convoluted barcoding strategy was employed; although, this strategy probably bore some similarity to those used Olson and Derda, the description in the supplementary, left many details up to the imagination.Overall, it is exciting to see mRNA display used in clever new ways, but it would also be nice to see this technique become much less specialized, empowering more labs to use RaPID in even craftier ways, such as enhancing PPI interactions, studying enzymatic reactions / catalysis, or evolving enzymes.