Azol(in)es, are important and common motifs in natural products and therapeutics. They are especially abundant in ribosomally synthesized and post-translationally modified peptide natural products (RiPPs) like cyanobactins and thiopeptides and even define an entire subclass of RiPPs dubbed “linear azole-containing peptides,” or LAPs. Discovered in 1976 by Asensio, Perez-diaz, Martinez and Baquero, the prototypic LAP, microcin B17, is a narrow-spectrum antibiotic isolated from E. coli that is effective against Pseudomonas and Enterobacteriaceae (minimum inhibitory concentration of 60 nM for E. Coli). Microcin B17 targets the bacterial topoisomerase, DNA gyrate, during DNA replication and works by interrupting the religation process producing double strand breaks in DNA. The four thiazoles and four oxazoles in microcin B17 are essential for this antibiotic activity and although a large body of work (with major contributions by Walsh, Kolter and Jung) has described their biosynthetic origins, until now, no structural evidence have corroborated these data.
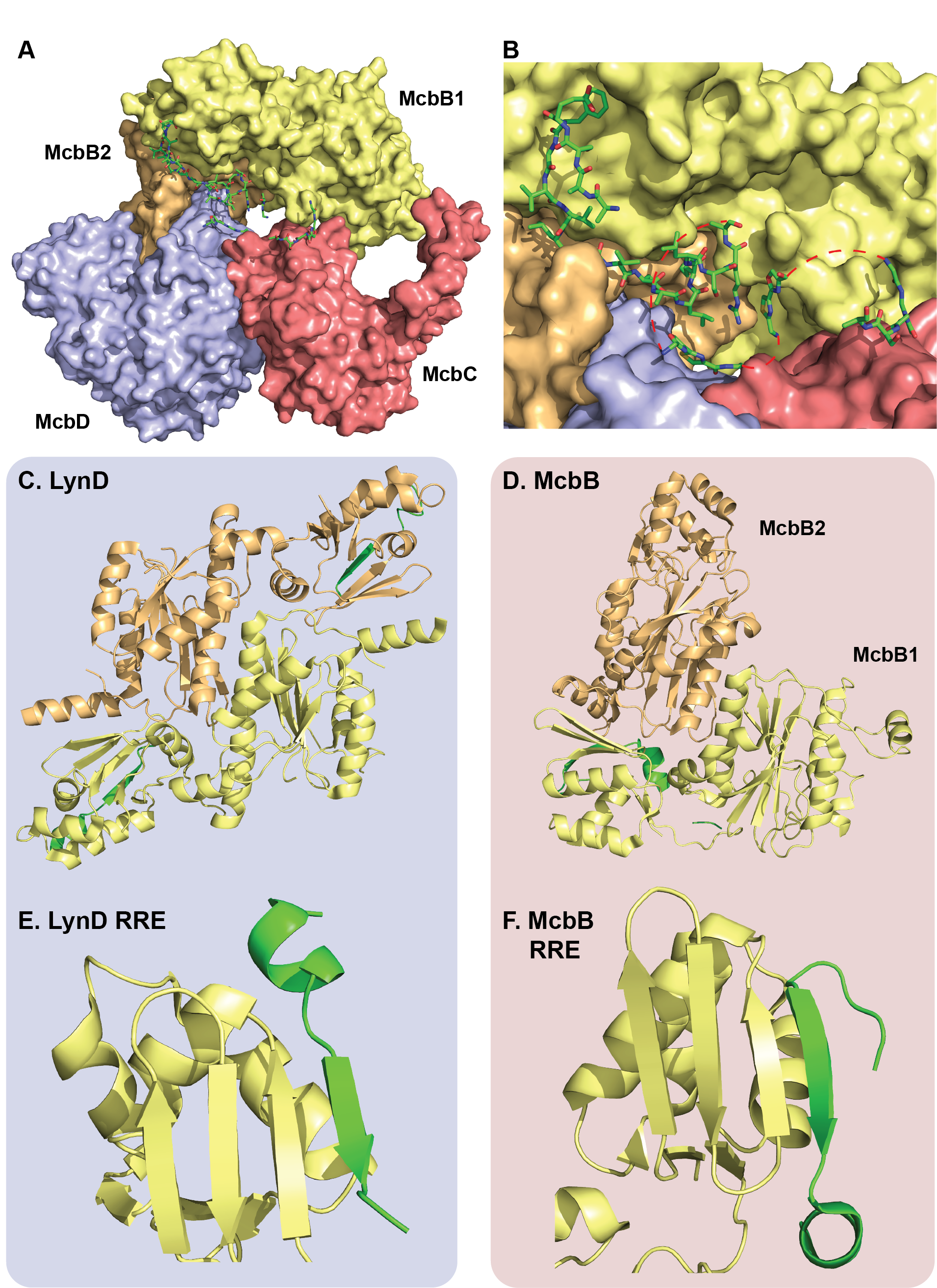
Recently, Severinov and coworkers captured multiple snapshots of the Microcin B17 synthetase at work (Figure 1A and B). A total of four different crystal structures, including three with peptide substrates and cofactors bound, confirm old hypotheses and provide unique insight. More specifically, these structures reveal how the complex uses a familiar leader peptide binding domain (the RiPP recognition element, RRE) to bind its substrate in a unique way. Taking a note from previous work, the authors leveraged the strong binding interactions between a hexahistidine-tagged McbA and its modifying enzymes (McbB, C and D) and were able to purify the entire complex at once in a single Ni-column purification. The first surprise came when the crystal structures revealed an octameric assembly of McbB4C2D2 in contrast to the expected 1:1:1:1 stoichiometry between McbA, B, C and D. The second unexpected feature was the structure of the leader peptide clamp which is made up of two conformationally distinct copies of McbB (Figure 1D). The leader peptide binds the RRE in one of the McbB copies in a similar manner to another heterocyclase LynD, however the asymmetric architecture of the McbB dimer is distinct from LynD (Figure 1C-F). The FxxxL motif in the McbA leader peptide was previously shown to be critically important for processing and the structure clearly shows two deep hydrophobic pockets in McbB that these residues occupy. Additional electrostatic interactions and a short helix just before the core peptide stabilize and orient the rest of the substrate towards the active sites. Interestingly, the orientation of the leader peptide is conserved between McbBCD and LynD (Figure 1E and F) suggesting the C-terminus of each core peptide approaches the active site first, however these enzymes modify their substrates in opposing directionality (LynD modifies in a C-to-N direction, while McbBCD modifies from N-to-C).
The cyclodehydratase McbD is structurally similar to LynD and is comprised of an ATP-binding pocket and an active site channel that is closed by a lid domain. The authors propose two possible hypotheses for cyclodehydration based on this structure. In one hypothesis (that we call the Cheerio theory, since many things in structural biology seem food-related), the active channel remains fairly static and the C-terminal end of the peptide substrate is threaded through (like string through a Cheerio), modifying from C-to-N. Although this mechanism is fairly consistent with the structural data, it doesn’t quite agree with previous biochemical data. A precursor bearing C-terminal β-gal can be correctly processed making it difficult to imagine how the C-terminus of this substrate can enter the channel. It is also inconsistent with the previously observed N-to-C modification directionality. A second “hinge” theory relies on a flexible lid domain, where an open-lid conformation allows the peptide to bind the active site, clamps down and then modifies the substrate in correct direction. Although this model would allow large C-terminal fusions to be processed, there is currently no evidence for the existence of multiple lid domain conformations.
Select alanine mutagenesis on the cyclodehydration domain emphasize residues P396, T148, E167 and a highly-coordinated water molecule all play important roles in cyclodehydration. The crystal structures also substantiate the proposed mechanism of the dehydrogenase, McbC. Critical residues K201 and Y202 abstract the α proton of the azoline, causing an E2 elimination of the β hydride to the flavin cofactor. The structure supports a stereoselective elimination. Lastly, the authors identify additional heterocycle binding motifs, so-called “LHW pocket” and “RF sandwich” which are suspected to transiently bind partially modified intermediates, thereby increasing the local concentration of the unmodified regions and promoting their modification.
These data complement decades of biochemical data and taken together, paint a detailed picture of how microcin B17 matures. The authors ultimately advocate for an overall mechanism where the precursor peptide is sequentially modified by shuttling back and forth between cyclization and dehydrogenase domains. While this is an enticing hypothesis, given the structural data collected, we remain cautiously optimistic. This model requires the precursor peptide to travel over 40 angstroms between each transformation and since no azoline-containing intermediate has ever been observed, this substrate repositioning would likely be fast and well-orchestrated and there is little structural evidence for these large conformational changes. The large complex (~240 kDa) may be well suited for CryoEM, which could allow for the identification of different conformations of the complex, perhaps with an open lid domain or with partially modified intermediates in one domain or the other (heterocyclization and/or dehydrogenase) to further support this shuttling hypothesis. Still, these structures present a variation of how RiPP-modifying enzymes use the RRE to bind their precursor peptides and provides a structural blueprint for LAP synthetases. Given the structural similarities to LynD questions arise over the reason for this distinct leader peptide binding orientation – why did the Mcb complex evolve into an asymmetric leader peptide dimer and why do LynD and the Mcb complex process their substrate is opposing directionalities? Additionally, the identification of heterocycle binding pockets intrigued our group. Azol(in)es are so ubiquitous in RiPPs, with natural products more often than not harboring numerous heterocycles, our lab wondered if these binding pockets may be the core peptide equivalents of the leader peptide’s RRE. Perhaps these binding pockets represent a conserved core binding strategy that serves as an allosteric substrate monitoring system that tells the enzyme when the peptide substrates have been sufficiently modified and triggers the release of a mature peptide. In-depth mutational, kinetic and binding analysis of partially modified intermediates (similar to what had been done for lanthipeptides) may shed light on these questions. Furthermore, the co-purification strategy used may be an efficient way to capture RiPP complexes in the future for biochemical and structural analysis.
One Response to “Grip it & RiPP it”
Dmitry Ghilarov
Haha, well done! Very pleased to read such a nice description and analysis of our work 🙂