This week, the lab has been reading about peptide therapeutics for malaria. Malaria is yet another infectious disease seeing a sharp rise in multi-drug resistant cases in recent years. Artemisinin and artemisinin combination therapies (ACTs) are the “go-to” treatment options for malaria, but resistant strains of Plasmodium falciparum, the malaria parasite, have been cropping up in Southeast Asia, South America, and elsewhere. Thus, new drugs with new modes of action are needed to target the malaria parasite within infected red blood cells before it can spread. One way to find new therapies is to look at how the human immune system works to combat the malaria parasite and, as it turns out, the immune system relies on peptides.
Lots of organisms deploy families of intrinsically structured, amphipathic peptides to protect against microbes. These naturally occurring antimicrobial peptides, or AMPs, associate with the typically charged surfaces of microbes and either disrupt the microbial membrane by means of insertion, or cross through it to inhibit intracellular targets. For some time now, AMPs have been investigated as potential therapeutics, but many of them can cause non-specific immune responses when translated into humans. Recent work by Lawrence et al., makes use of an AMP-like domain from human platelet factor 4, or PF4, as a starting point for the development of a potential new peptide therapeutic against malaria. PF4 is a small chemokine protein, released by platelets in response to infections, such as malaria. The idea here is to develop the C-term 14-amino acids, as a non-immunogenic AMP for malaria. The N-term otherwise induces chemokine cascades that could be detrimental and lead to conditions such as cerebral malaria. A second reason for developing the C-terminal domain over the whole protein is the inability of the protein to target infected red blood cells (RBCs) without a chemokine receptor known as DARC. There is an allele that encodes for a lack of DARC in the membrane of RBCs and this allele is common in Africa where the deadliest strain of malaria, Plasmodium falciparum, accounted for 99% of the estimated malaria cases in 2016.
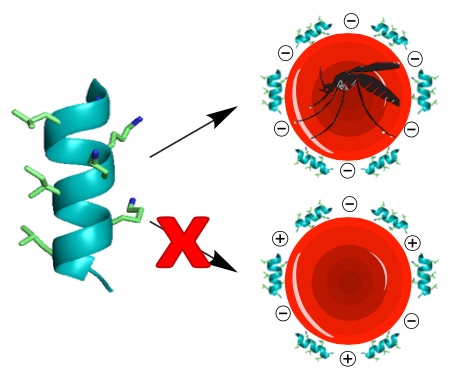
To optimize the PF4 peptide, the authors minimized the C-terminal domain to its last 14 amino acids, which they employed as its monomer (PF4P57-70) and as a dimer (PF4PD), as well as fashioning it into a cyclic peptide dimer (cPF4PD). Circular dichroism and human serum stability studies confirmed that the PF4PD and cPF4PD constructs exhibited improved stability and a-helicity. The group then looked into the mechanism of these peptides by means of liposome models of infected and uninfected RBC membranes. These studies showed that the peptides acted through disruption of the substantially more charged organellular membranes of the parasite inside infected RBCs and specifically the digestive vacuole of the parasite. They also found evidence of parasite DNA fragmentation that could lead to parasite death. Lastly, they looked at the peptides’ ability to specifically target infected RBCs and the parasitic organelles. Again, they could attribute the observed specificity to the peptides’ higher affinity for more negatively charged membranes of infected RBCs and parasitic organelles. This is an important finding in the sense that it suggests that it may be possible to design peptides capable of targeting the altered infected cells, and that clues to the specific design principles may already be present in nature.
Although the paper provides a thorough mechanistic investigation of the PF4-derived peptides, it does little to situate it within the broader context of AMPs. Given the breadth of research in this area, it would be useful to know what specifically distinguishes this human made AMP from the much more well-known variants in microbes and insects and how those differences might affect immunogenicity. Along those lines, although the stated purpose is at least in part to optimize for immunogenicity, there are no specific studies aimed at this feature, however they do suggest in vivo testing and intestinal permeability as key next steps. One point that the authors bring up at the end and that our lab found interesting is the idea of using these peptides to carry another molecule into the infected RBCs. Such a cargo might target a specific pathway in the parasite and augment the potency of the peptide; it recalls the use of more common cell-penetrating peptides in other applications but in this case the cell-penetrating peptide comes with its own activity and is capable contributing to killing the parasite. Regardless, the idea of taking lessons in medicinal chemistry from the immune system seems worth pursuing further.