Recently we delved into the literature on new and improved arylomycin antibiotics, in particular, this recent report in Nature by researchers at Genentech. Arylomycins are a class of macrocyclic peptides known to inhibit the bacterial type I signal peptidase pathway of Gram-positive bacteria viaLepB inhibition. It was previously thought that arylomycins had extremely narrow-spectrum activity against Streptococcus pneumoniae and a handful of soil bacteria. Work by Romesberg published in 2010, however, demonstrated that mutations in the bacterial strains resulting in reduced activity of the natural arylomycin compounds was due to destabilizing interactions via hydrogen bonding to the lipophilic tail with the mutated residues. This finding has breathed new life into the arylomycins, which have now been modified to reveal their much broader spectrum activity against a wide array of Gram-negative ESKAPE pathogens. Due to continued emergence of multi-drug resistant (MDR) Gram-negative bacteria, the need for new classes of drugs with novel mechanisms of action are imperative. No new class of antibiotics for the purpose of treating MDR Gram-negative pathogens has been approved in over fifty years.
Romesberg and Baran reported a new and improved synthetic route to the arylomycin macrocycle by way of an oxidative aryl-aryl (C-H activation) reaction. Synthetic access to the arylomycin core has seemingly been streamlined, opening the door for facile and divergent chemical derivatization of this class of antibiotic.
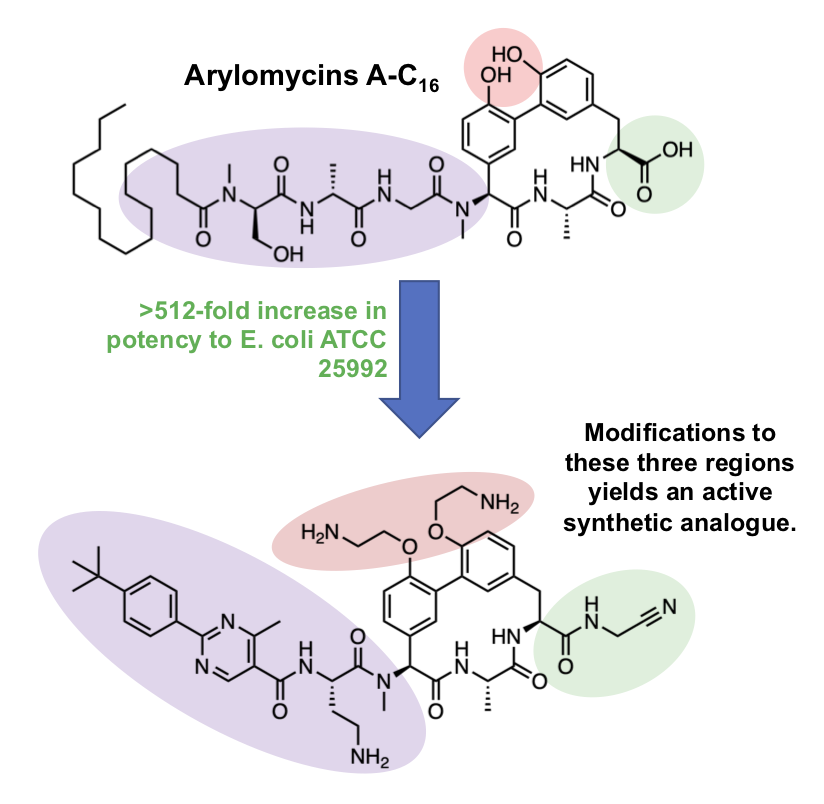 Still more recently, the Genentech group reported a new compound, G0775, an arylomycin analogue, that exhibits potent antibacterial activity against a variety of ESKAPE group pathogens. While the Baran chemistry is streamlined, the Genentech group exploited a Suzuki route developed by Zhu. The authors chose to make modifications to the N-terminal lipopeptide tail, phenolic oxygens, and the C-terminal carboxylic acid. Prior SAR has shown that modifications to the first two may result in improved activity and that conservation of the macrocycle is important for binding. A large flat aromatic tail, unable to form destabilizing hydrogen bonds (as observed in the natural arylomycins), results in improved activity, as does ethylamine substitution of the two phenolic oxygens. Most interestingly, the C-terminal modifications provided major improvements in activity. The natural compound’s C-terminal carboxylic acid binds via the Ser-Lys catalytic dyad of LepB, however, G0775’s aminoacetonitrile “electrophilic warhead” is capable of forming a covalent bond with the Lys146 side chain of LepB. Together, these modifications result in drastically improved potency (low µM) against E. coli and other ESKAPE pathogens compared to the natural compound (>64 µM).
Still more recently, the Genentech group reported a new compound, G0775, an arylomycin analogue, that exhibits potent antibacterial activity against a variety of ESKAPE group pathogens. While the Baran chemistry is streamlined, the Genentech group exploited a Suzuki route developed by Zhu. The authors chose to make modifications to the N-terminal lipopeptide tail, phenolic oxygens, and the C-terminal carboxylic acid. Prior SAR has shown that modifications to the first two may result in improved activity and that conservation of the macrocycle is important for binding. A large flat aromatic tail, unable to form destabilizing hydrogen bonds (as observed in the natural arylomycins), results in improved activity, as does ethylamine substitution of the two phenolic oxygens. Most interestingly, the C-terminal modifications provided major improvements in activity. The natural compound’s C-terminal carboxylic acid binds via the Ser-Lys catalytic dyad of LepB, however, G0775’s aminoacetonitrile “electrophilic warhead” is capable of forming a covalent bond with the Lys146 side chain of LepB. Together, these modifications result in drastically improved potency (low µM) against E. coli and other ESKAPE pathogens compared to the natural compound (>64 µM).
G0775 is shown to permeate the outer cell wall via a porin-independent, most likely self-promoted uptake mechanism. This result of broad spectrum antibiotic activity has also recently been correlated to a compound’s tendency to hold additional positive charge, an artifact of cell wall penetrance that ultimately manifests itself in amines (Hergenrother, 2010). A minimal frequency of resistance against ESKAPE group pathogens is also reported in this manuscript, underscoring G0775’s potential against MDR bacteria. Interested in G0775’s clinical potential, several in vivo studies were carried out to determine its ability to treat systemic and pulmonary bacterial infections. Not only did G0775 exhibit drastically improved activity against ESKAPE bacteria at lower dosages than current clinical antibiotics (Ciprofloxacin), but G0775 lacked mammalian cell toxicity as well.
In total, this work paints a picture of a compound whose broad spectrum activity was rescued first by a better understanding of resistance mutations and second by improved penetrance into Gram-negative outer layers. It makes us wonder how many more active molecules, natural products in particular, might be rescued by improving their penetrance and/or editing away components responsible for resistance. Because natural products typically must be synthesized by bacteria without killing the producer, similar design elements may have been used more broadly in other natural products to keep them from being active against the producer. Editing natural products for activity and/or penetrance may be in the future like turning the ‘safety’ off a whole new arsenal of weapons against infectious disease.